




Esophagus Cancer
Esophageal Cancer: Esophageal cancer, which is 3 t...

Primary hyperaldosteronism is the secretion of excessive aldosterone from the adrenal cortex and suppression of plasma renin activity (PRA), resulting in hypertension - hypokalemia.
There are 2 subtypes of Primary Hyperaldosteronism:
These constitute 95% of all cases. Although the treatment of the first is surgical, the treatment of the second is medical; does not respond to surgery.
All adenomas are always unilateral, and most have an average diameter of 1.8 cm. In terms of clinical features, it is most common between the ages of 30 and 60. The male/female ratio is 2/3.
The usual clinical symptoms are muscle weakness, polydipsia (drinking too much water), nocturia (frequent urination at night) and headache. For a definitive diagnosis, plasma renin activity (PRA) and plasma aldosterone concentration (PAC) values should be monitored. If the results of hormonal measurements cannot help the diagnosis, additional tests can be performed. If aldosterone production cannot be suppressed with a diet containing high Na+ or PRA cannot be stimulated with a diet containing low Na+, the diagnosis of primary aldosteronism can be approached. Excision of an aldosterone-producing adenoma and retained adrenal is curative. However, surgery is not very urgent. The disease can be treated with spironolactone or amiloride in mild cases, often without time limits, but the side effects of spironolactone at high doses (impotence, gynecomastia and postural hypotension) are not well tolerated. Weakness as a result of hypokalemia justifies the choice of surgery for recovery. Laparoscopic unilateral total adrenalectomy is the standard intervention option for well-localized adenomas. When the adenoma is removed in surgery, the blood pressure response is excellent in approximately 70% of cases. The other 30% requires appropriate anti-hypertensive treatment.
Pheochromocytoma occurs in the adrenal medulla or related chromaffin tissue anywhere in the body; tumors that secrete dopamine, epinephrine or norepinephrine (or all 3) – sometimes including vasoactive peptides. These result in persistent or episodic hypertension or other symptoms of excessive catecholamine secretion.
The classic symptoms are episodic hypertension with palpitations followed by flushing, palpitations, headache, excessive sweating (perspiration), irritability and anxiety. An important feature is the triad of palpitation, headache and sweating that occur simultaneously. symptoms
Pheochromocytoma in adults is often benign. There are multiple tumors in 10% of cases. Malignant tumors are more common in children. For screening, determinations of vanillmandelic acid (VMA) and metanephrine in urine are useful for the purpose.
10% of all Pheochromocytoma cases
it is malignant
It is bilateral
located outside the adrenal
seen in children
it is familial
shows recurrence
It is associated with MEN syndromes
It occurs with a cardiovascular crisis.
Complications of pheochromocytoma are mostly sequelae of hypertension. If a biochemical diagnosis is made, treatment with alpha adrenergic blocking agents should be started immediately. The definitive treatment for pheochromocytoma is excision. The favorite surgical procedure in the 21st century is laparoscopic adrenalectomy.
(Cushing's Disease and Cushing Syndrome)
Cushing's syndrome is the result of excessive cortisol and corticosterone secretion.
70% of the cases are pituitary-dependent Cushing's disease (=pituitaryCushing Syndrome). The case is ectopic Cushing's syndrome. ACTH-independent Cushing syndrome cases.
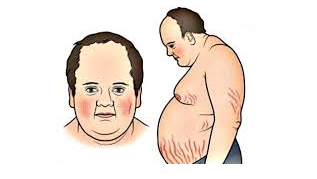
It constitutes 20% among; Most of these are unilateral cortisol-secreting adrenal adenomas or carcinomas.
Women are dominant with a ratio of 10:1. Although the age range of occurrence is between childhood and the 8th decade, the most common incidence is in the 3rd and 4th decades.
The classic definition of Cushing syndrome includes truncal obesity, hirsutism, moon face, acne, buffalo back, red citria, hypertension and diabetes. The most notable symptoms are fatigue and depression.
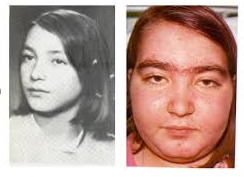
Diagnosis is difficult. For this reason, free cortisol in 24-hour urine is one of the most discriminative and definitive tests available.
Nelson Syndrome occurs in approximately 20% of cases after adrenalectomy. This is the result of excessive secretion of the pituitary.
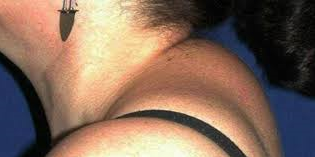
Cushing's syndrome can be treated by surgery or irradiation of the pituitary, adrenals, or both, or by trying to modify adrenal hormone synthesis.
Medical treatment: Temporary control of Cushing's disease is possible with metyrapone and aminoglutethimide.
Irradiation of pituitary adenomas is effective; However, it is necessary to wait 8-18 months to achieve a certain effect, which is too long for a rapidly progressing disease. Recurrences are common after radiotherapy.
Adrenalectomy: Patients with severe Cushing's syndrome are not good candidates for surgery. Post-operative complications are common; wound infection, hemorrhage, peptic ulceration, and pulmonary embolism are at the top of the list.
The safest treatment for Cushing syndrome caused by bilateral hyperplasia is total bilateral adrenalectomy.
Unilateral laparoscopic adrenalectomy is the favorite surgical treatment method for patients with adrenal adenoma.
Resection is the treatment of choice for malignant tumors. Treatment for benign adenomas is unilateral adrenalectomy, but these are rarely bilateral.
After total adrenalectomy, lifelong corticosteroid maintenance therapy is required.
The most common cause of primary adrenal insufficiency is the deterioration of the adrenal gland cortex layer.
Secondary adrenal insufficiency: It is a condition that occurs secondary to anomalies in the hypothalamus and pituitary.
The most common cause is long-term administration of glucocorticoids. (iatrogenic)
Schmidt syndrome: Autoimmune adrenal insufficiency may coexist with other endocrine autoimmune diseases. It is most often accompanied by hypothyroidism due to autoimmune thyroiditis.
DIAGNOSIS: There is hyperpigmentation.
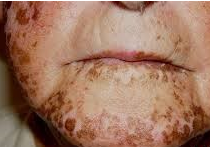
Findings such as pain in the upper abdomen and flanks, abdominal rigidity, vomiting, confusion and hypotension may be detected; in this case, secondary spontaneous adrenal insufficiency due to adrenal bleeding or adrenal vein thrombosis should be considered.
Intraadrenal hemorrhage may develop due to sepsis, anticoagulant treatment or coagulopathy.
Waterhouse-Friedrichsen syndrome: It is an acute adrenal hemorrhage secondary to severe sepsis. It most commonly occurs as a result of meningococcal sepsis.
To confirm the diagnosis, blood cortisol, glucose, sodium, potassium, blood urea nitrogen and creatinine levels should be checked and a complete blood count should be performed.
Blood table of the patient in adrenal crisis;
TREATMENT: If acute adrenal crisis is suspected iv. Hydrocortisone should be given. Intravenous access is established with dextrose and balanced salt solutions, 3 * 100 mg for 2 days. Hydrocortisone is continued. The dose is gradually reduced for the next few days, decreasing to a maintenance dose of 12-15mg/m2/day and continuing at this dose.
If there is primary adrenal insufficiency, mineralocorticoid should be started when hydrocortisone reaches the maintenance dose. (fluorocortisone 0.1-0.2 mg/day)
The most common cause of virilization is congenital adrenal hyperplasia. Feminization, which occurs as a result of an adrenal disease, is almost always due to adrenal tumors, the majority of which are malignant. The only known treatment is excision.
Clinical signs of virilization include any of the following: early sexual maturity, acne, increased hirsutism, male appearance, male hairstyle and hair loss, and increased or decreased libido in men. The main symptoms in women are atrophy of secondary sex characteristics, decreased libido, hirsutism and acne. Rapid growth occurs in prepubertal women. Tumor-induced virilization is accompanied by hypertension.
Feminizing and masculinizing tumors are large and easily seen on CT or MRI-scans.
Treatment consists of excision of the tumor. Complications of the surgery are relatively rare. Tumors are rarely bilateral or ectopic.
These can reach large sizes, but are rarely malignant. They can usually be decompressed and removed abdominally.
These lesions, which are detected during a diagnostic attempt other than adrenal masses, are found incidentally on CAT or MRI-scan and rarely have clinical significance for surgeons. Excision; It should be reserved for tumors larger than 6 cm or growing during the observation period.